2017 年的新发现提高了我们对血压调控机制的认识。在免疫机制、肠道菌群的作用、血管周围脂肪和炎症对脉管系统的负面影响、以及肾素-血管进展素-醛固酮系统中少见的基因变异方面的关键研究,为我们认识高血压的发病机制提供了新的见解。来自加拿大蒙特利尔 McGill 大学高血压科、医学部以及医学研究所的专家们对 2017 年高血压病发病机制方面的新进展进行了总结,发表在最新一期的 Nature Review Nephrology 杂志上。
关键进展
1. 血清和 T 细胞中的糖皮质激素调节蛋白激酶(SGK1)信号传导,通过一种涉及到上调 Na+/K+/2Cl-转运子 1(NKCC1)和极化 T 辅助细胞 17(Th17)表型的机制,会引起盐诱导性血压升高和靶器官损害。
2. 高盐饮食改变了人类和小鼠的肠道菌群,导致乳酸杆菌数量的减少,Th17 细胞数量的增多,血压升高。
3. 在小鼠中,γδT 细胞参与了血管紧张素Ⅱ诱导的高血压和内皮细胞功能紊乱,也可能在人类高血压和终末期脏器功能损害中发挥作用。
4.AngⅡ的 1a 型受体(AGTR1A)和骨桥蛋白介导了周围脂肪组织中巨噬细胞极化到炎症表型的过程,导致了炎症和潜在有助于主动脉瘤形成。
5. 包括 APLN 基因和 RENBP 基因在内的 7 个肾素-血管紧张素-醛固酮系统(RAAS)相关基因的少见变异与血压的盐敏感性有关。
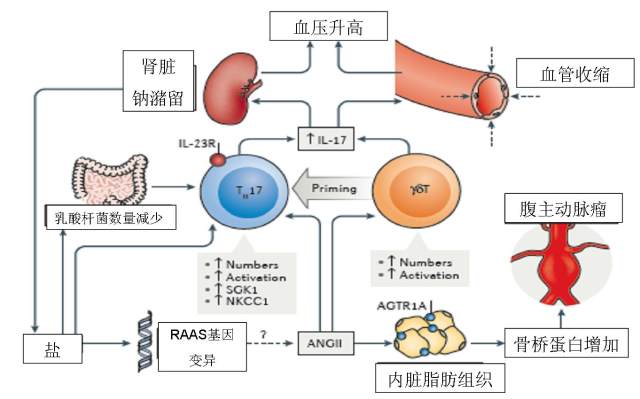
图 1 高血压和主动脉瘤形成的新机制
盐(NaCl)通过提高 NKCC1 活性的血清和 SGK1 依赖途径从而诱导 Th17 细胞活化。高盐摄入同样也导致肠道菌群中乳酸杆菌的数量减少,导致 Th17 细胞的刺激。血管紧张素Ⅱ(AngⅡ)刺激γδT 细胞,其反过来又刺激免疫其它细胞,包括 Th17 细胞。活化的 Th17 细胞和γδT 细胞产生 IL-17,这将刺激肾脏钠潴留和血管收缩,导致血压升高。较少见的 RAAS 基因变异可能通过 AngⅡ参与机制增加盐的敏感性。在血管周围的内脏脂肪组织中,AngⅡ与 AngⅡ的 1a 型受体(AGTR1A)结合会导致致炎症的骨桥蛋白产生增加,这些有助于腹主动脉瘤的形成和进展。
