近期的 Kidney International 杂志上刊登了一个不同寻常病因的慢性肾脏病(CKD)病例,我们一起来学习一下吧!
病例介绍
一位 65 岁的非洲加勒比裔的老年男性,因肾功能恶化被转诊至肾脏科诊所。他的 eGFR 在过去的一年内从 37 ml/min/1.73m2降至 23 ml/min/1.73m2。他的既往史包括:慢性背痛、贝尔式面瘫、高血压病和痛风。他有慢性肾脏病家族史,女儿最近被诊断为肾脏的结节病。临床体格检查无特殊。
尿液分析显示微量蛋白尿,没有血尿。肾脏超声显示双肾无梗阻。感染和免疫介导的肾脏病方面的筛查以及多发性骨髓瘤的筛查均阴性。进行了肾活检以明确他肾功能恶化的病因。组织学和偏光显微镜下的结果如图 1 所示。
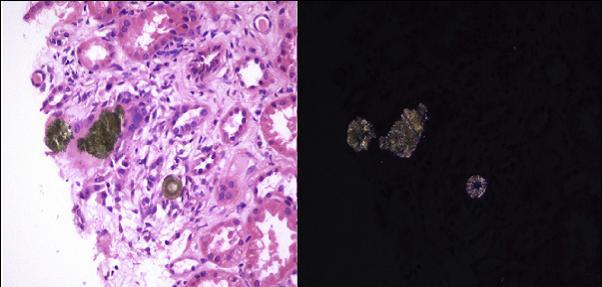
图 1 肾活检的光镜和偏振光图片
你的诊断是什么?
诊断:腺嘌呤磷酸核糖基转移酶(APRT)缺乏症伴结晶性肾病
病例讨论
肾活检显示肾小管内绿色/棕色结晶样物质,上皮细胞引出多核巨细胞反应。在偏光显微镜下,晶体清晰可见折射力。还可见慢性肾实质损害伴慢性炎症细胞浸润。
考虑到 2,8-二羟基腺嘌呤(DHA)晶体的典型特点,疑似「腺嘌呤磷酸核糖基转移酶(APRT)缺乏症」的诊断,随后通过实验室检查红细胞 APRT 酶活性完全缺乏得以证实。
APRT 是一种从腺嘌呤中回收 5』-腺苷酸的嘌呤补救酶。在 APRT 缺乏症中,腺嘌呤被黄嘌呤脱氢酶(也称为黄嘌呤氧化酶)转化成 DHA 所替代。DHA 经过肾脏清除,但是大部分不溶解于尿液,沉积在肾小管里,导致梗阻性肾病和晶体性肾病。
APRT 缺乏症是一种常染色体隐性遗传性疾病,表现为 APRT 酶活性非常低或缺乏。经典的报道是在婴儿中导致肾结石,现在在成人中被诊断为 APRT 缺乏症的病例也日益增多。任何年龄都可以有临床表现,可归因于尿路结石或「DHA 肾病」。
虽然是遗传性疾病,但是液体摄入量和嘌呤消耗量被认为是疾病表型变异的影响因素。引用报道的发病率为 1/50000~100000 人,因为对该病的认识不够,并且通常表现不典型,这一数字很可能低估了真实的发病率。
APRT 缺乏症的诊断可以通过在肾脏结石或尿中结晶中确定 DHA 而做出。这常常需要尿液离心和偏振光下沉积物的显微分析。随后通过测定红细胞 APRT 酶的活性证实了该诊断。肾活检对于诊断该病不是必须的,但是在我们的病例中,肾活检是识别明确该病症的一种手段。由于突变鉴定的异质性,遗传分析不是诊断。
如果不治疗的话,APRT 缺乏症可导致终末期肾病,即使在移植肾中也会复发。虽然目前没有治愈的可能性,但是 APRT 缺乏症很容易治疗。别嘌醇,通过抑制黄嘌呤脱氢酶,防止了腺嘌呤转化为 DHA。经过别嘌醇 300 mg qd 治疗后,6 个月后患者的 eGFR 改善至 30 ml/min/1.73m2。
虽然很少见,但是 APRT 缺乏症却很容易治疗。当面对未知类型的肾结石或不明原因 CKD 的患者时,尤其是尿常规中活动性尿沉渣不多的患者时,需要考虑 APRT 缺乏症的可能性。
